Medical device regulations, classification and submissions
Read time: 6 mins
Read the highlights (click here)
Medical devices play a crucial role in the diagnosis, prevention, monitoring, and treatment of diseases. Unlike drugs or biologics, a medical device can vary from the simple, which poses little or no risk to the user (for example, a toothbrush), to the life-sustaining (for example, a pacemaker). The technology and design can rely on any combination of mechanical, electronic, software or chemical/biochemical action to achieve their purpose.1,2 Registration processes (that is, to reach regulatory clearance or approval) for medical devices vary greatly across jurisdictions.
Classification and data requirements
Due to the wide variety of medical devices, these products are regulated on a risk-based classification system. In Canada and the EU, devices are grouped into four different classes.1 In the US, they are divided into three groups.2 Generally, the higher the risk of the medical device, the higher the medical device classification. With a higher classification come more stringent data requirements to demonstrate the device’s safety, effectiveness, and performance.
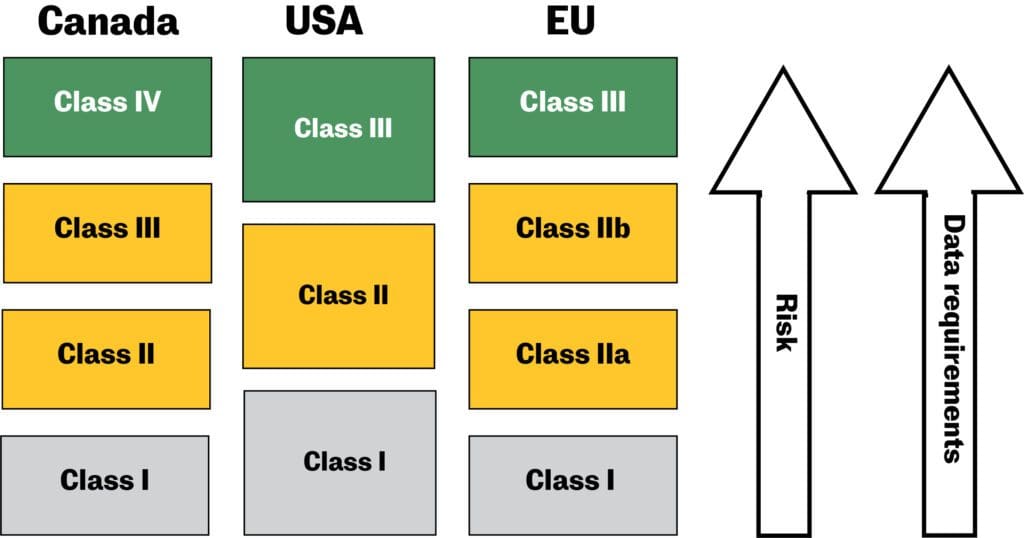
Procedures to classify medical devices
The classification procedures in Canada and the EU are quite similar―manufacturers must classify their medical devices according to the rules and criteria set out in the relevant medical device regulations (Canada) and directives (EU), as indicated in Table 1.
In the U.S., the classifications and ancillary information relating to medical device regulations are published in US Code of Federal Regulations (CFR), Title 21, Parts 862–892. These were introduced in 1976 when the U.S. Food & Drug Administration (FDA) mandated expert advisory panels (classification panels) to consider the different types of medical devices on the market based on the intended user, intended use, the risk-to-benefit ratio and the reliability of the device.
In the U.S., manufacturers of a new medical device can determine their classification by comparing its intended use and technological features to those that have already been classified. However, if the device is technologically innovative with no existing predicate device, it will automatically be classified as class III. Manufacturers of class III medical devices may petition the FDA for a lower classification via a “de novo” process, based on supporting objective scientific evidence as to the device’s safety and effectiveness.3-6
Regulations or directives related to medical device classification
| Jurisdiction | Applicable medical device regulations/ directives |
| Canadai | Medical Devices Regulations (SOR/98–282). Schedule 1. Classification rules for medical devices.7
|
| U.S. | Code of Federal Regulations (CFR), Title 21, Parts 862–8925 |
| EUi |
Changes in the new directive:
|
Going to market: Medical device licenses and registration
Canada
Manufacturers, distributors and importers who wish to sell a medical device must obtain an establishment license for class I devices. For class II, III or IV medical devices, the company must obtain a medical device license issued by Health Canada. To do so, they must submit a device license application and include a certificate demonstrating compliance to ISO 13485:2003. The application for class II devices is administrative in nature. Applications for class III devices are based on the submission of summary documents, and those for class IV devices are based on extensive data, including all study reports, quality plan, risk assessments and so on.4
Health Canada has established an emergency use pathway for devices by way of an interim order to allow devices that are approved in other jurisdictions to be imported or sold in Canada. Interim orders are used in response to public health emergencies and detail the categories of devices that are eligible. The process with Health Canada depends on the class of device being imported or sold and must remain in compliance with Canadian Medical Device Regulations. The use authorization is only valid for the duration of the interim order.8
U.S.
Most class I (and also a few class II) medical devices are exempt from registration requirements as specified in the CFR Parts 862–892. If registration is exempted, the manufacturer must register their establishment with the FDA and comply with the applicable Quality System Regulation (QSR), labelling and medical device reporting requirements. For other class I and II devices that are subject to Premarket Notification, a 510(k) application must be submitted for FDA clearance. If the device is classified as a class III device, the manufacturer must go through the Premarket Approval (PMA) procedure, or the de novo procedure to reclassify the product in a lower-risk category before placing it on the market.5,6
The FDA has also established its Breakthrough Device Program market pathway, which exists for devices deemed to treat life-threatening or debilitating diseases with a novel technology. It is not an alternative for the classifications and processes cited above; however, it offers a partnership with the FDA wherein it is more involved throughout the proceedings to ensure a more efficient and complete submission. To qualify, submitters must demonstrate that there is no alternative to their technology currently available in the market and their device is gravely needed for patient care. The initial step of this process is to apply for this designation with the FDA.9
The FDA also has an emergency use authorization that can be accessed when overall public health is deemed to be under threat. This allows unapproved medical devices to be used in emergency situations. Generally, a request can be submitted to the FDA for review and, if appropriate, an approval letter will be granted. This authorization is only valid for the length of the emergency and for the devices types that the agency has identified as eligible.10
EU
On May 26, 2021, the European Union (EU) Medical Device Regulation (MDR) (2017/745) replaced the EU Medical Device Directive (MDD). The new MDR introduced a major change to the regulatory framework in the EU. The modernization of the regulatory system brought several changes to the information required for devices and their regulatory documentation. Not all changes apply to all medical devices—the changes to the regulatory documentation of the MDR vary from product to product. Annex I, General Safety and Performance Requirements outlines the new conditions that need to be addressed by devices currently CE marked under the MDD. Existing products must be re-certified under the new MDR by May 26, 2024.
![]()
For low-risk medical devices (Class I non-sterile non-measuring), the manufacturer may make a declaration of conformity with the General Safety and Performance Requirements (GSPRs), based on a self-assessment without the involvement of a notified body.‡ For other medical devices, a notified body’s involvement is required.11,12,13
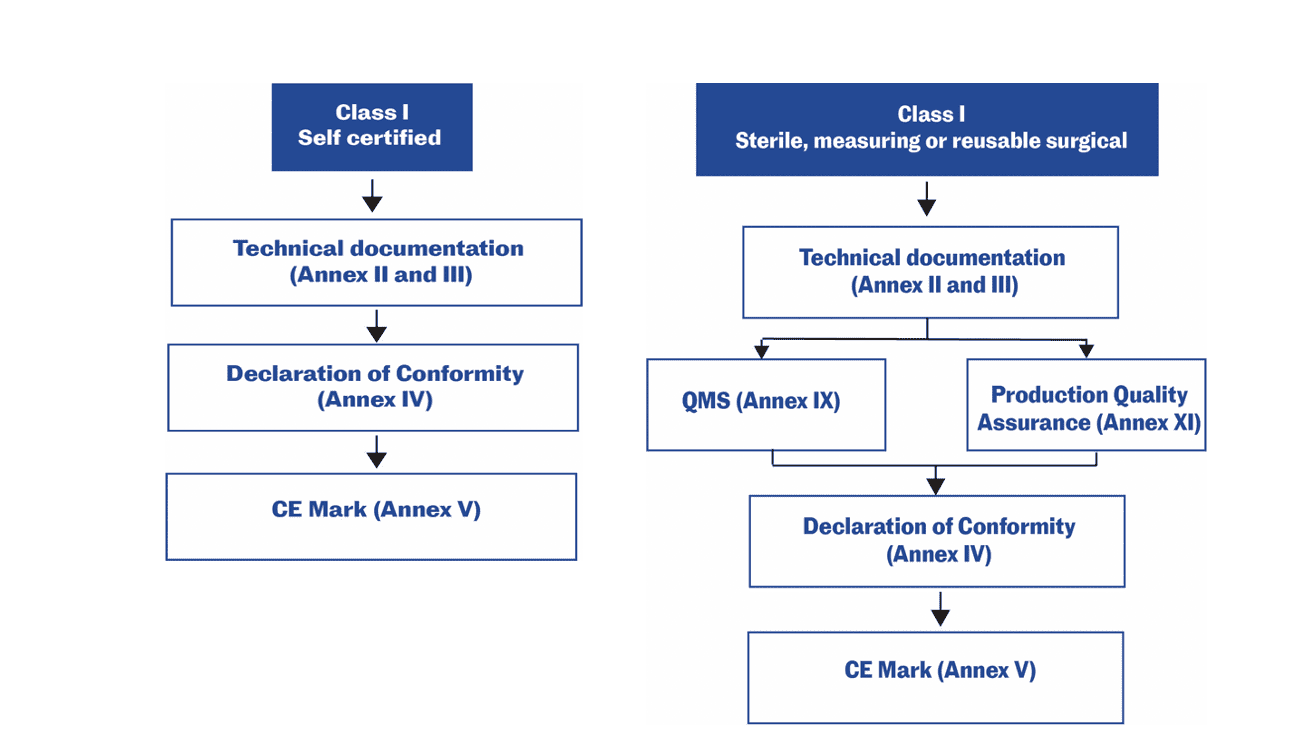
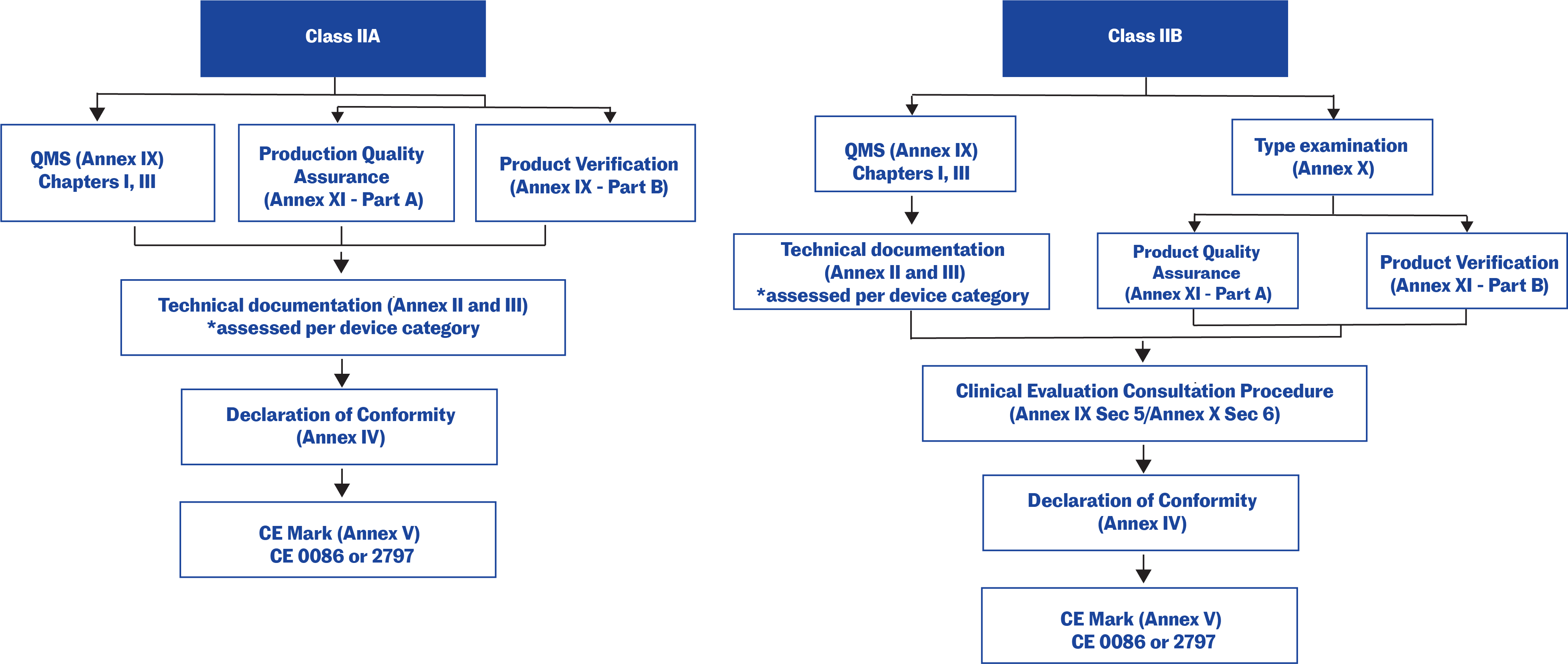
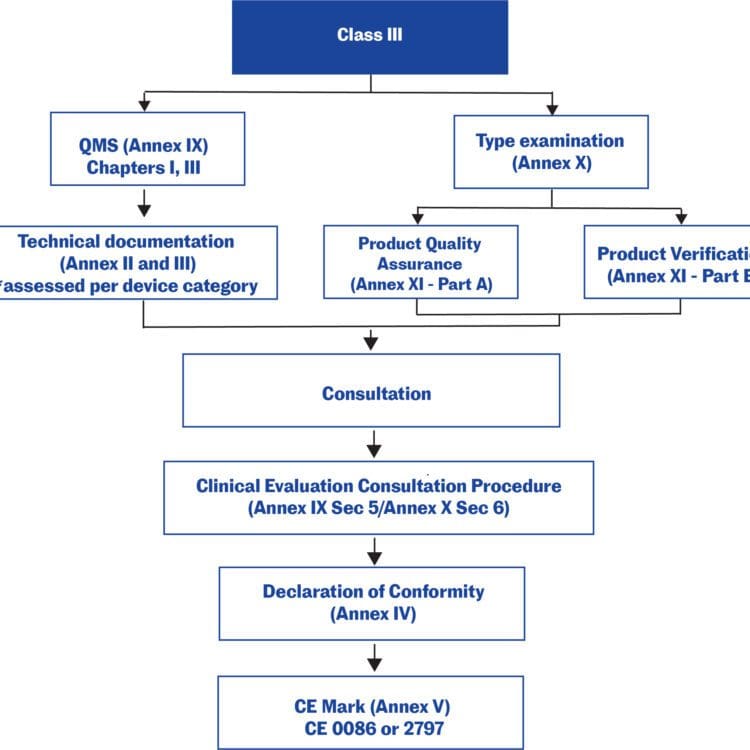
Technical documentation in the EU
Technical documentation that provides evidence of conformity with the General Safety and Performance Requirements (GSPRs) set out in Annex I of the Regulation (EU) 2017/745. The technical documentation should include relevant performance data, procedures, standards, labelling, certifications by a notified body and/or the manufacturer’s declaration of conformity. The technical documentation must be maintained at the disposal of the Competent Authorities (national authorities in the EU Member States) for a period of five years after manufacture of the last product. Requests from Competent Authorities to view the technical documentation will usually be triggered by problems in the market.
iIf more than one rule applies, a medical device must be assigned the highest class that applies under the various rules.
iiA notified body is certification organization that the national authority (the Competent Authority) of an EU Member State designates to carry out one or more of the conformity assessment procedures described in the annex(es) of the EU Directives. It must be qualified to perform all the functions set out in any annex for which it is designated.14
Summary: For medical devices, regulatory clearance/approval/licensing procedures vary across jurisdictions, with more stringent requirements applied depending on the level of risk associated with the product.
Disclaimer
The information presented in these articles is intended to outline the general processes, principles and concepts of the healthcare product development lifecycle. Since regulatory requirements are ever-changing, it is current only as of the date of publication and not intended to provide detailed instructions for product development. Every healthcare product is unique and therefore so is its associated product development lifecycle. Specific advice should be sought from a qualified healthcare or other appropriate professional.
Updated: February 15, 2021
References
- Classification Of Medical Devices And Their Routes To CE Marking. (November 2020). Retrieved November 8, 2021 from https://support.ce-check.eu/hc/en-us/articles/360008712879-Classification-Of-Medical-Devices-And-Their-Routes-To-CE-Marking
- Classify Your Medical Device. (February 7, 2020). Retrieved November 9, 2021 from https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
- Tobin, J.J. & Walsh, G. (2008). Chapter 9. Medical devices. In Medical product regulatory affairs. Pharmaceuticals, diagnostics, medical devices. Weinheim: Wiley-VCH Verlag GmbH & Co.
- Guidance Document – Guidance on the Risk-based Classification System for Non-In Vitro Diagnostic Devices (non-IVDDs). (April 23, 2015). Retrieved November 3, 2021, from https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/application-information/guidance-documents/guidance-document-guidance-risk-based-classification-system-non-vitro-diagnostic.html
- United States Code. Title 21, Parts 862–892 (April 1, 2020). Retrieved November 3, 2021, from http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm
- U.S. Food and Drug Administration. (2021, October 10). Evaluation of Automatic Class III Designation (De Novo) Summaries. Retrieved November 3, 2021, from http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ucm232269.htm
- Classification Rules for Medical Devices. (2021, June 23). Retrieved November 3, 2021, from https://laws-lois.justice.gc.ca/eng/regulations/SOR-98-282/page-13.html#h-1022100
- Interim order respecting the importation and sale of medical devices for use in relation to COVID-19. (2020, June 19). Retrieved November 3, 2021, from https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/interim-order-importation-sale-medical-devices-covid-19.html
- Breakthrough Devices Program. (2021, January 5). Retrieved November 3, 2021, from https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program
- Emergency Use Authorization of Medical Products and Related Authorities. (2017, January). Retrieved November 3, 2021, from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities
- EurLex. (1993, June 14). Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. Retrieved November 3, 2021, from http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:31993L0042:EN:HTML
- European Commission. (2009, June 5). Implementation of Directive 2007/47/EC amending Directives 90/385/EEC, 93/42/EEC and 98/8/EC. Retrieved July 16, 2012, from https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:247:0021:0055:en:PDF
- Tobin, J.J. & Walsh, G. (2008). Chapter 10. Authorisation of medical devices. In Medical product regulatory affairs. Pharmaceuticals, diagnostics, medical devices. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA.
- Notified bodies. Retrieved November 9, 2021 from https://ec.europa.eu/growth/single-market/goods/building-blocks/notified-bodies_en